általánosság
A Pompe-betegség (vagy a II. Típusú glikogén-tárolási betegség ) egy ritka örökletes betegség, amelyet a glikogén túlzott felhalmozódása jellemez.

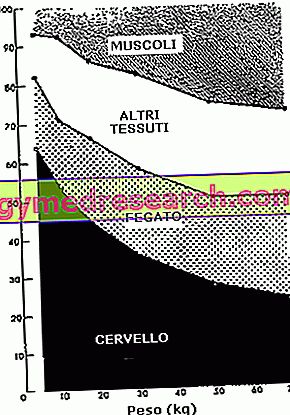
A glükózpolimer megsemmisítéséből eredő károkat elsősorban a májban, a szívizomban és a vázizomokban fejezik ki, amelyek fokozatosan gyengülnek .
A Pompe-betegséget genetikai aberráció okozza, amely a lizoszómákban (intracelluláris organellákban) a sav-alfa-1, 4-glükozidáz enzim ( GAA vagy maltázsav ) aktivitásának hibáját eredményezi. különböző típusú molekulák lebomlása).
A II. Típusú glikogenózis infantilis formája a cardiomegalia súlyos formájához vezet, amely a második életévben a cardiorespiratory dekompenzációval halált okozhat. A késői megjelenésű Pompe-betegségben azonban a tünetek az izomra korlátozódnak, és az infantilis változathoz képest e progresszív myopathia progressziója viszonylag jóindulatú.
Mi
Glikogén: kulcsfontosságú pontok
- A glikogén az állati sejtekben a fő tartalék poliszacharid.
- A glikogén glükóz alegységekből áll, és tárolási forrásként felhalmozódik a máj citoszoljába (ahol különösen gazdag), a vázizomzatból és kisebb mértékben a veséből.
- A glikogént a szervezet a vércukor támogatására használják éhgyomorra, általában étkezés és éjszaka között.
Mi a glikogenózis?
A glikogenózis ritka betegségek egy csoportja, amelyet egy vagy több glikogén anyagcserében részt vevő enzim örökletes hiánya okoz. Ezeket a körülményeket a különböző szervekben lévő glikogénfelesleg feleslege erősíti .
Pompe-betegség - II. Típusú glikogenózis: mi ez?
A Pompe-betegség örökletes, krónikus és progresszív kóros állapot, amelyet a glikogén felhalmozódása jellemez a sejtekben, vagy néhány intracelluláris organellumban, például a lizoszómákban.
A glikogén lebomlásáért felelős enzim hibája miatt az utóbbi felhalmozódik és károsítja a szív, a lábak és a karok és a légzés izmait.
Pompe-betegség: szinonimák és terminológia
A 2. típusú glikogén tárolási betegségnek is nevezik, a Pompe-betegség a lizoszomális tárolási rendellenességek csoportjába tartozik.
Ennek a patológiának a szinonimái számosak, és magukban foglalják:
- Sav-alfa-1, 4-glükozidáz-hiány ;
- Glikogenózis savas maltázhiány miatt ;
- 2. típusú glikogén tárolási betegség (más néven glikogén tárolási betegség II . Típus, glikogén tárolási betegség, 2. típus vagy GSD, 2. típus );
- 2. típusú glikogén tárolási betegség .
okai
Pompe-betegség - II. Típusú glikogenózis: mi az oka?
A Pompe-betegség egy lizoszomális tárolási rendellenesség, amelyet a lizoszomális enzim - alfa-1, 4-sav-glükozidáz (vagy savas maltáz ) - veleszületett hiánya okoz, amely általában felelős a glikogén elhelyezéséért .
A II. Típusú glikogén tárolási betegségben ez az enzimhiba a következőket eredményezi:
- Zavarok, amelyek a szervezetnek a glikogénként tárolt glükóz helytelen használata miatt nem képesek;
- A glikogén felhalmozódása által okozott változások a lizoszómákban (intra-lizoszomális) vagy ezeken kívül (extra-lizoszomális).
A Pompe-kór alapjában lévő enzimhiány valójában mindenütt jelen van, de az ebből eredő károsodás mindenekelőtt bizonyos szervekben (beleértve a szív- és / vagy a vázizomzatot) fejeződik ki.
Hogyan terjed a Pompe-betegség?
- A Pompe-betegség bázisánál a GAA-gén néhány mutációja található a 17q23-as kromoszómán .
- A génhibát autoszomális recesszív módon továbbítják: ez azt jelenti, hogy a szülőknek mind a "mutáció egészséges" hordozói, mind a terhességek esetén 25% -os kockázata lesz a Pompe-betegségben szenvedő gyermekek 50% -os valószínűségének. egészséges gyermekekkel és 25% -kal egészséges gyermekekkel, akik nem hordoznak.
- A II. Típusú glikogenózis patogenetikai mutációi heterogének, ezért a betegség klinikai megjelenése változó. Néhány aberráció gyakoribb, mint mások. Mindenesetre az eredmény a lizoszomális enzim normális aktivitásának károsodása, amelyből a glikogén felhalmozódását követi.
Tünetek és szövődmények
Pompe-betegség - II. Típusú glikogenózis: hogyan nyilvánul meg?
A Pompe-betegség komplex és heterogén klinikai megjelenést mutat, amely a sav-alfa-1, 4-glükozidáz különbségének mértékétől függ, valószínűleg az ugyanazon enzimet kódoló GAA-gén különböző mutációival függ össze.
Jellemzően a II. Típusú glikogén tárolási betegséget az izomszövet (myocardium, vázizom és légzés) és a máj változó károsodása jellemzi.
Általánosságban elmondható, hogy a lizoszóma-aktivitás károsodása fokozatosan az izomerő hiányához vezet.
Gyerekes forma
A Pompe-betegség infantilis formája az élet 3 hónapja előtt következik be:
- A csontváz izomzatának hipotoniaja;
- A nyelv bővítése;
- A szopás és a nyelés nehézsége;
- Hipertrofikus kardiomiopátia;
- Progresszív hepatomegalia;
- Mentális retardáció.
A Pompe-betegség fertőző formái a legsúlyosabbak, mivel:
- Cardiomegalia ( szívdiagnózis, amely a szívelégtelenség következtében halált okozhat);
- Súlyos izomgyengeség, motoros készségek késleltetett megszerzése vagy regressziója.
Kezelés hiányában a Pompe-betegség fertőző formáiban a kardiorespirációs dekompenzáció következtében a halál a két életév előtt következik be.
Késői vagy felnőtt formája
A késői formákban - azaz az élet első évét követő bármely korban - a II. Típusú glikogenózis serdülőkorán vagy "felnőttkori" megjelenésénél a progresszió lassú, és a következmények kevésbé kedvezőek, mint a csecsemők, mivel a szív általában meg van takarítva.
A Pompe-betegség főként az alsó végtagoktól kezdődő gyengülés hatására érinti az izmokat, és légzési problémákat okoz .
Idővel valójában ez a myopathia vezethet ahhoz, hogy önállóan ne tudjon ambulálni, míg a szellőztető kapacitás fokozatosan veszélyeztetett, ami légzési elégtelenséghez vezet. Kezelés nélkül a II. Típusú glikogén tárolási betegségben szenvedő betegeknek vagy segített szellőztetéssel vagy tracheostómiával kell rendelkezniük.
jegyzet
A klinikai kép tekintetében a csecsemő és a késői forma Pompe-betegség két szélsőségét képviseli. E két változat között a köztes formák széles skálája létezik.
diagnózis
Hogyan készül a 2. típusú glikogenózis diagnózisa?
A Pompe-betegség diagnózisa a klinikai megfigyelésen és az enzimhiány bizonyítékán alapul. A gyanú megerősítéséhez különösen a GAA enzimatikus aktivitását tenyésztett bőr fibroblasztokban, limfocitákban vagy izombiopsziás mintában mérjük:
- A Pompe-betegség infantilis formájával rendelkező gyermekeknél a sav-alfa-1, 4-glükozidáz aktivitása gyakorlatilag hiányzik;
- A késői formában a maradék enzimaktivitás különböző szintjei vannak.
Továbbá, a genetikai (beleértve a prenatális) elemzést a GAA génben lévő mutációk keresésével lehet elvégezni. A korai diagnózis segíthet a betegek életminőségének javításában.
Pompe-betegség: differenciáldiagnózis
A késői forma differenciáldiagnosztikája a myopathia egyéb okainak köszönhető.
A Pompe-betegség csecsemőformáját elsősorban az alábbiak szerint kell megkülönböztetni:
- Werdnig-Hoffman-betegség (1. típusú SMA);
- Hipertrofikus kardiomiopátia (idiopátiás vagy metabolikus).
Pompe-betegség: lehetséges a prenatális diagnózis?
A Pompe-betegség prenatális diagnózisa a következő módon végezhető:
- Az enzimatikus aktivitás mérése a friss chorionos foltokban a 12. terhességi héten;
- Keressük meg a magzat sejtjein, a magzati folyadékból vett GAA génmutációkat a terhesség 15. hetében.
Jelenleg ez a betegség nem tartozik az újszülött szűrés által lefedett betegségek közé (jegyzet: eddig néhány régióban, köztük Toszkána és Veneto) kísérleti kísérletet folytatnak.
kezelés
Pompe-betegség: milyen kezelések állnak rendelkezésre?
A 2. típusú glikogén tárolási betegségre jelenleg rendelkezésre álló kezelési lehetőségeket enzimhelyettesítő terápia képviseli: 2006 óta az Európai Unió engedélyezte a ritka gyógyszer forgalomba hozatalát Pompe-betegségben szenvedő betegek kezelésére, \ t alfa-alglükozidáznak nevezik. Gyakorlatilag a terápia a biotechnológiai úton előállított enzim periodikus intravénás beadását jelenti (két hetente egyszer) rekombináns úton.
Az enzimpótló terápia hatékonyabb, ha a betegség korai szakaszában adják be. Az ezzel a megközelítéssel kapható eredményeket illetően a rekombináns humán GAA-val történő enzimhelyettesítő terápia nagyon hatékony a csecsemő Pompe-betegségben, mivel jelentősen meghosszabbítja a túlélést és csökkenti a kardiomiopátiát. Néhány késői formában ez a terápiás megközelítés a betegség stabilizálódásához vezethet.
Egyéb lehetséges beavatkozások
A Pompe-betegség kezelése tüneti kezelést és néhány olyan gyógyszert tartalmazhat, amely javíthatja az enzimhelyettesítő kezelés hatásait.
A génterápia kiaknázásának lehetősége a 2-es típusú glikogén tárolásban még tanulmányozás alatt áll.