általánosság
Az Apert-szindróma egy olyan genetikai rendellenesség, amely a brachycephaly nevű craniosynostosis formájáért felelős, és az ujjak és lábujjak szindróma nevű rendellenessége.

Az Apert-szindróma újszülöttenként minden 68 000-88 000-ben megfigyelhető az FGFR2-gén specifikus mutációjának köszönhetően, amelynek feladata a koponya-varratok fúziójának szabályozása és az ujjak és lábujjak kialakulása.
Az Apert-szindróma diagnosztizálásához, a fizikai vizsgálathoz, a történelemhez, a koponya és az ujjak és lábujjak radiológiai értékeléséhez, és végül egy genetikai vizsgálathoz elengedhetetlen.
Jelenleg az Apert-szindrómában szenvedők csak tüneti kezelésekre támaszkodhatnak, azaz enyhítik a tüneteket és elkerülik a legsúlyosabb szövődményeket.
A koponya varratok és azok fúziójának rövid áttekintése
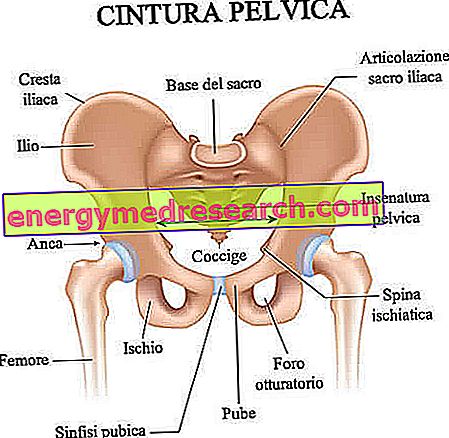
A koponya varratok a rostos ízületek, amelyek együttesen biztosítják a koponyatükör csontjait (azaz a frontális, időbeli, parietális és occipitalis csontokat).
Normál körülmények között a koponya varratok fúziós folyamata a szülés utáni időszakban történik, 1-2 évesen kezdődően, bizonyos ízületi elemeknél, és mások számára a 20 éves küszöbértékben. Ez a hosszú és ritmikus fúziós folyamat lehetővé teszi az agy növekedését és fejlődését.
Mi az Apert szindróma?
Az Apert-szindróma az egyik legfontosabb genetikai betegség, amely craniostenózist (vagy craniosynostosisot ) okoz, azaz az úgynevezett koponya varratok korai fúzióját.
Az Apert-szindróma azonban nemcsak a craniostenosishoz való kötődésével, hanem azzal is, hogy egy bizonyos fokú syndactylyhoz kötődik, ez a veleszületett rendellenesség, amelyet egy vagy több kéz vagy ujj összekapcsolása jellemez. láb.
A craniostenosis és a syndactyly egyidejű kiváltásának lehetősége az Apert-szindrómát az acrocephalosyndactyly példaként teszi; az orvostudományban az acrocephalosyndactyly olyan genetikai állapot, amely egyesíti a koponya specifikus rendellenességeit ("acrocephalus" jelentése "hegyes fej") egy vagy több ujj vagy lábujj fúziójához.
Milyen következményei vannak a koponya varratok korai fúziójának?
Ha, mint az Apert-szindróma és más kapcsolódó betegségek esetében, a koponya-varratok fúziója a prenatális, perinatális (*) vagy korai csecsemőkori, agyi szervek, például az agy, a kisagy és az agytörzs, valamint a szervek alatt történik. értelme van, mivel a szemek növekedési és formai változásokon mennek keresztül.
* Megjegyzés: a "perinatális élet" a 27. terhességi hét és a születést követő első 28 nap közötti időszak.
Epidemiológia: milyen gyakori az Apert-szindróma?
A statisztikák szerint egy 65 000-88 ezer fő között Apert-szindrómával születne.
Tudtad, hogy ...
Genetikai betegségek, amelyek, mint Apert-szindróma, craniosynostózisokat okoznak, 150 körül vannak.
Ezek közül kiemelkedik az Apert-szindróma mellett a Crouzon-szindróma, a Pfeiffer- szindróma és a Saethre-Chotzen-szindróma .
okai
Az Apert-szindróma az FGFR2-gén specifikus mutációjának köszönhető, amely a 10. kromoszómán található.
A legtöbb esetben az embrionális fejlődés során a fent említett mutációt teljesen spontán és pontos okok nélkül szerezzük be - azaz a sperma megtermékenyítése után megkezdődött a tojás és az embriogenezis; ritkábban örökletes - vagyis egy vagy mindkét szülő továbbítja.
kíváncsiság
Az Apert-szindrómát okozó szerzett mutáció a de novo mutáció egyik példája, azaz az "új mutáció, amely teljesen mentes az öröklődő természetből".
Mi okozza az Apert-szindrómához kapcsolódó gén mutációját?
Előfeltétel: a humán kromoszómákon jelenlévő gének olyan DNS-szekvenciák, amelyeknek alapvető feladata az élethez nélkülözhetetlen biológiai folyamatokban alapvető fehérjék előállítása, beleértve a sejtek növekedését és replikációját.
Ha az mutánsoktól mentes (ezért egészséges emberben), az Apert-szindrómában szerepet játszó FGFR2-gén megfelelő mennyiségben termel egy receptorfehérjét, a Fibroblaszt növekedési faktor receptort 2, amely elengedhetetlen a koponyahegyek olvadási idejének jelöléséhez. és az ujjak és lábujjak szétválasztásának szabályozására (más szóval azt jelzi, hogy a megfelelő idő a koponya varratok fúziójára szolgál, és szabályozza az ujjak és lábujjak kialakulását).
Ha ezután az Apert-szindróma jelenlétében megfigyelt mutációt hajtja végre, az FGFR2-gén hiperaktív, és a fent említett receptorfehérjét olyan nagy mennyiségben termeli, hogy a koponya-varratok fúziójához kapcsolódó időzítés megváltozik (gyorsabb) és az elválasztási folyamatok a következők: az ujjak és a lábujjak nem fordulnak elő helyesen.
Ki a leginkább veszélyeztetett?
Az Apert-szindróma megszerzett esetét illetően az FGFR2-gén mutációját indukáló tényezők jelenleg nem teljesen egyértelműek.
A kérdéssel kapcsolatos kutatás még folyamatban van.
Az Apert-szindróma egy autoszomális domináns betegség
Megérteni ...
Minden emberi gén két példányban van jelen, az allélok, az anyai eredetű és az apai eredetűek.
Az Apert-szindróma az autoszomális domináns betegség minden jellemzőjével rendelkezik.
A genetikai betegség az autoszomális domináns, ha a gén csak egy példányának mutációja elegendő ahhoz, hogy megnyilvánuljon.
Tünetek és szövődmények
Amint azt az elején megállapítottuk, az Apert-szindróma két klinikai tünetgel jár: craniostenosis (vagy craniosynostosis) és syndactyly.
craniostenosis
Különböző típusú craniosynostosis van; a különbözõ típusok megkülönböztetése a korai fúziónak kitett koponya- varratok.
Az Apert-szindróma esetében a normálisan jelenlévő craniostenosis típusát brachycephaly- nak hívják. Koronás craniosynostosis néven is ismert, a brachycephaly a koronális varratok korai fúziója, vagyis a frontális csont és a parietális csontok között futó koponya varratokból eredő koponya anomália (célszerű a koponya varratok alakját tekinteni).

A brachycephaly jelensége a következő hatásokkal jár:
- A koponya előrehaladásának elmulasztása előre és hátra, ami viszont az agy abnormális növekedéséhez vezet oldalirányban és felfelé;
- Magas és kiemelkedő homlok kialakítása és a koponya lapos háttere. Az Apert-szindrómás egyén fejének általános megjelenése egy hosszú fej, amely függőlegesen alakult ki;
- Az intrakraniális nyomás többé-kevésbé jelentős növekedése (azaz az agy nyomása, amelyet a koponya csontjai ellen gyakorolnak). Különösen ha súlyos, az intrakraniális nyomás növekedése olyan tüneteket okozhat, mint:
- Folyamatos fejfájás;
- Látási problémák;
- hányás;
- ingerlékenység;
- Hallási problémák;
- Légzőszervi problémák;
- A mentális állapot változásai;
- Papilla.
- A szellemi fejlődés hiányosságai, amelyek az intellektus csökkent kapacitását jelzik ( alacsony IQ ). Az értelem csökkent képessége a páciensektől óriási mértékben változik, és függ a koponya varratok korai fúziója által kiváltott malformáció súlyosságától;
- Arc-rendellenességek, beleértve:
- Lapos vagy homorú arc (az arc központi csontjainak növekedésének hiánya miatt);
- Puffy, kiálló és széles szemmel; sekély szemcsatlakozók és szokatlanul elválasztott szemek (a szemcsatlakozók hipertelorizmusa);
- Csőr orr;
- Alacsony fejlett állkapocs, prognosztikával kombinálva;
- Zsúfolt fogak (a fejletlen állkapocs miatt);
- Fülek alacsonyabb magasságban, mint a normál.
Syndactyliát
Az Apert-szindróma hordozóiban a szindikátus a kézben szinte mindig és a lábakban ritkábban figyelhető meg, mint a kezekben.

Az Apert-szindrómás egyén kezében a szindikta tipikus jellemzői 4:
- Rövid hüvelykujj sugárirányú eltéréssel (vagyis anomálisan a rádiumra orientálva, az alkar két csontjának egyike);
- Komplex szinkronizálja a mutatóujját, a középső ujját és a gyűrűs ujját. A komplex szindikátussal az orvosok az ujjak rendellenes fúzióját jelentik, amely nemcsak a lágy szöveteket (a bőrt), hanem a csontszöveteket (a phalangeket) is érinti;
- Sinfalangismo . Az orvostechnikai kifejezés azt jelzi, hogy az ujjak interfangangális ízületeinek anomális fúziója van (az interphalangealis ízületek a falanx és a falanx közötti ízületi elemek);
- Egyszerűen szinkronizálódik a negyedik és az ötödik ujj között (azaz a gyűrűs ujj és a kisujj között). Egyszerű szindikcióval a szakértők az ujjak rendellenes fúziójára utalnak, amely csak a lágy szöveteket (a bőrt) érinti.
A SYNDACTYLOUS SZÖVETSÉGE A NYITOTT SZINDÓMÁBAN: A 3 TÍPUSOK
A hüvelykujj rendellenességének súlyossága alapján (a négy jellemző előtt) az Apert-szindrómában résztvevő szakemberek megkülönböztetik a fokozottan súlyosabb szindaktilia 3 típusát:
- Az I. típus (a legkevésbé súlyos) egybeesik a hüvelykujj minimális anomáliájával, amely teljesen független az indextől.
Egyéb anomáliák: az index, a középső és a gyűrűs ujj egy komplex szinaktikusan és jelen lévő sinfalangizmussal fuzionálódik a disztális interphangangális ízületekért; a gyűrűs ujj és a kis ujj között egyszerű és hiányos szinkronizálódik (a hiányos szinaktika azt jelenti, hogy a két ujj közötti fúzió részleges).
Egyéb információ: ez a leggyakoribb típus.
- A II. Típusú (köztes gravitáció) jellemzője a hüvelykujj sugárirányúabb eltérése az előző esethez képest, és az azonos hüvelykujj és a mutatóujj közötti szinaktikus elv (a hüvelykujj és a mutatóujj között hiányos szinaktika van).
Egyéb anomáliák: az index, a középső és a gyűrű egy komplex szindikátus főszereplője, a distalis sinfalangismo-val kombinálva; a gyűrűs ujj és a kisujj között egy egyszerű és hiányos szindikátus van.
Egyéb információ: ez a második leggyakoribb típus.
- A III. Típus (a legsúlyosabb) a hüvelykujj, amely az indexben össze van kötve, nem csak a lágy szövetekben, hanem a csontszövetekben is.
Egyéb anomáliák: minden ujj olvad össze, annyira, hogy szinte lehetetlen felismerni őket; csak egy szög van; ha az első 4 ujj között szinaktikusan összetett, a gyűrűs ujj és a kis ujj között egyszerű és hiányos (mint a többi típusnál).
Egyéb információ: ez a legritkább típus.
Az Apert-szindróma egyéb lehetséges tünetei és jelei
Bizonyos esetekben az Apert-szindróma a craniostenosishoz és a syndactylyhez való kötődésen kívül a következő: polydactyly (azaz egy extra ujj jelenléte a kézben vagy lábban), hallásvesztés, ismétlődő fülfertőzések és paranasalis sinusok, hyperhidrosis, zsíros bőr, súlyos pattanások, a szemöldökhártya hiánya, a nyaki csigolyák fúziója, obstruktív alvási apnoe szindróma és / vagy szájpadlás.
szövődmények
Az Apert-szindróma szövődményei mindenekelőtt azok a súlyos következmények, amelyeket a craniosynostosis az agy fejlődésére és a szellemi kapacitásra, valamint a szindikált kezek funkcionális kapacitására gyakorolhat.
Mikor lehet észrevenni az Apert-szindrómát?
Az Apert-szindrómából eredő koponya- és digitális rendellenességek általában születéskor nyilvánvalóak, ezért a diagnózis és a kezelés tervezése azonnali.
diagnózis
Általában az Apert-szindróma diagnosztizálásához vezető vizsgálatok lefolyása a fizikai vizsgálat és az anamnézis után kezdődik; ezért folytatódik egy sor radiológiai vizsgálat a fejen (röntgen a fejen, CT a fejen és / vagy mágneses rezonancia a fejen) és a kezek és végül a lábak; végül egy genetikai próbával végződik.
Fizikai vizsgálat és kórtörténet
A fizikai vizsgálat és az anamnézis lényegében a beteg által feltárt tünetek gondos vizsgálatában áll.
Az Apert-szindróma összefüggésében a diagnosztikai eljárás ezen pillanataiban az orvos megvizsgálja a craniostenózist és a szindikátust, valamint azok pontos jellemzőit.
A kezek és lábak fejének és ujjának radiológiai vizsgálata
Az Apert-szindróma összefüggésében:
- A röntgenfej tesztek szükségesek ahhoz, hogy az orvos megerősítse a koronális varratok (koronális craniostenosis vagy brachycephaly) korai fúzióját; továbbá lehetővé teszik, hogy megbecsülje a koponya-agyi rendellenességek súlyosságát.
- Másrészről az ujjakon és a lábujjakon végzett radiológiai vizsgálatok nem annyira alapvetőek a syndactyly megerősítéséhez (ehhez elegendő a vizuális vizsgálat), hanem inkább az interdigitális fúziók konnotációinak részletes ismertetése (a szindikaktikus jelenlét típusa, szintje). egyesülése stb.).
Genetikai teszt
A DNS-elemzés célja a kritikus gének mutációinak detektálása.
Az Apert-szindróma összefüggésében ez a megerősítő diagnosztikai teszt, mivel a szóban forgó genetikai betegségre jellemző FGFR2-mutációt világítja meg.
terápia
Jelenleg az Apert-szindrómaért felelős mutáció nem gyógyul ; azonban a ritka genetikai betegség hordozói még javíthatják állapotukat, mivel rendelkeznek több tüneti kezeléssel, amelyek a tünetek enyhítésére irányulnak, elhalasztják az elkerülhetetlen szövődményeket, és végül elhárítják azokat, amelyek elkerülhetők.
Tudtad, hogy ...
Annak érdekében, hogy egy olyan betegségből, mint Aperté-szindróma helyreálljon, meg kell szüntetni az embrionális fázis genetikai mutációját, hogy a koponyaház és az ujjak és a lábujjak megfelelően növekedjenek.
Tüneti terápia: mit tartalmaz?
Az Apert-szindróma jelenlétében alkalmazott minden tüneti kezelés alapja:
- A brachycephalia sebészeti kezelése és következményei érettebb korban, pl
- Syndactyly sebészeti kezelése .
Ezért a páciens által bemutatott tünetek függvényében a felelős orvos kiegészítheti a fent említett kezeléseket:
- Terápiás terv, amely néha meglehetősen invazív az obstruktív apnoe szindróma ellen;
- Terápiás terv a paranasalis sinus fertőzések megismétlődése ellen;
- Egy sebészeti kezelési terv, melynek célja a fülfertőzések megelőzése (ha ezek ismétlődnek, ha nem visszatérő, de szórványos, elegendő antibiotikum terápia);
- Sebészeti kezelési terv a lehetséges anomáliák megoldására, mint például a szájpadlás, a nyaki csigolyák fúziója stb.
A BRACHICEPHALIA KIRÁLYI KARBANTARTÁSA
Az Apert-szindróma hordozója esetében a brachycephaly sebészeti kezelése a következőket tartalmazza:
- Az első beavatkozás fiatal korban (az életévben), amelynek célja a falu koronális varratok elválasztása a vártnál korábban . Ha ez a beavatkozás sikeres, az agynak megfelelő a hely a növekedéshez, és csökken az intellektuális problémák kockázata.
- Egy második beavatkozás 4 és 12 év között, amelynek célja, hogy normális megjelenést biztosítson az arcnak, ami (ahogy az olvasó emlékszik) lapos, ha nem is konkáv.
A szóban forgó művelet magában foglalja az arc néhány csontjának bemetszését és azok áthelyezését olyan struktúra szerint, amely legalább részben tükrözi a normálisságot.
- Harmadik esetleges beavatkozás a gyermekkori években, azzal a céllal, hogy megszüntesse vagy legalábbis csökkentse a fehérorosz szemészeti szemléletet .
A SZYNDRIL KEZELÉS SZERVIZ KEZELÉSE
A syndactyly műtéti kezelése az interdigitális fúzió jellemzőinek függvényében változik (ezért a típusától függ).

Ez azt jelenti, hogy az Apert-szindrómás egyénre érvényes beavatkozás nem egyformán érvényes egy másik, azonos genetikai betegségben szenvedő egyénre (csak akkor, ha a syndactyla típusa azonos).
Miután ezt az alapvető szempontot tisztázták, a meglévő sebészeti megközelítés minden típusa ugyanaz, és az ujjak összegyűjtésével együtt egyesül, annak érdekében, hogy bizonyos funkciókat biztosítson a kezeknek.
Általában a syndactyla kezelése két szakaszból áll:
- 1 lépés: az első interdigitális tér szabadítása (a hüvelykujj és az index közötti tér) és a negyedik interdigitális tér (a gyűrűs ujj és a kis ujj közötti tér);
- 2 lépés: a második és a harmadik interdigitális tér szabadítása (az index és a közep közötti tér és a középső és a gyűrű közötti tér).
prognózis
A craniostenosis megfelelő kezelése nélkül az Apert-szindróma biztosan negatív prognózist mutat, a beteg még súlyos szellemi problémákkal küzd; ezzel szemben a megfelelő beavatkozás gyakorlása során a szóban forgó genetikai betegség jóindulatú prognózist élvezhet, nem biztos, hogy negatív, a beteg normális vagy majdnem normális IQ-t mutat.
Tudtad, hogy ...
A családban növekvő Apert-szindrómás 10 gyermekenként négy közülük normál IQ-t fejleszt.
megelőzés
Az Apert-szindróma lehetetlen a megelőzés feltétele.