általánosság
Az Alport-szindróma egy ritka örökletes genetikai rendellenesség, amely fokozatos veseelégtelenséget, hallásvesztést és szembetegséget okoz.

Az Alport-szindróma oka az, hogy a vesék, a belső fül és a szem megfelelő működéséhez szükséges fehérje előállításában szerepet játszó gének mutációját képezik.
Az Alport-szindróma diagnózisa a fizikai vizsgálaton, a kórtörténeten, a vesebiopszián és a genetikai teszten alapul.
Jelenleg az Alport-szindrómában szenvedők csak tüneti kezelésekre támaszkodhatnak, azaz enyhítik a tüneteket és elhalasztják a szövődményeket.
Mi az Alport szindróma?
Az Alport-szindróma egy ritka genetikai rendellenesség, amely súlyos veseproblémákat, halláskárosodást és szembetegségeket okoz azoknál, akik ezeket hordozzák.
Az Alport-szindróma öröklött állapot, ahol az „örökletes” kifejezés „egy vagy mindkét szülő által továbbított”.
Epidemiológia: milyen gyakori az Alport-szindróma?
A statisztikák szerint minden 50.000-en Alport-szindrómával születik.
Szinonimák
Az Alport-szindróma öröklött természetéből adódóan, és vesebetegségéből adódóan, az orvostudomány területén is ismert örökletes nefritisz .
okai
Az Alport-szindróma egy olyan mutációnak tulajdonítható, amely egy rendellenes változás, a COL4A3, COL4A4 és COL4A5 betűkkel nevezett három emberi gén közül egyben vagy többben .
Hol tartózkodnak az Alport-szindrómáért felelős gének?
Míg a COL4A3 és a COL4A4 gének esetében a lokalizáció mind a humán genom 2. kromoszómája, a COL4A5 gén az emberi genom X kromoszómáján van .
A 2. kromoszóma egy autoszomális kromoszóma ; az X kromoszóma viszont egy nemi kromoszóma, amely egy kromoszóma, amelyen az egyén neme függ.
A mutációk hatása
Előfeltétel: a humán kromoszómákon jelenlévő gének olyan DNS-szekvenciák, amelyeknek alapvető feladata az élethez nélkülözhetetlen biológiai folyamatokban alapvető fehérjék előállítása, beleértve a sejtek növekedését és replikációját.
Ha a mutációktól mentesek (ezért egészséges emberben), az Alport-szindrómához kapcsolódó 3 gén alapvető fehérje komponenst hoz létre a vesében, a belső fülben előforduló IV. Típusú kollagén megfelelő előállításához, feldolgozásához és végső strukturálásához . és a szemekben .
Ahelyett, hogy a mutációk áldozatai lettek volna, az Alport-szindrómához kapcsolódó 3 gén elvesztette a fent említett IV. Típusú kollagén megvalósításához nélkülözhetetlen fehérje-komponens létrehozásának képességét, és ez magában foglalja a vesék, a belső fül és a szem szöveti megváltozását. e szervek által okozott zavarok.
Tudtad, hogy ...
A IV. Típusú kollagén bevonása Alport szindrómába kötőszöveti betegséggé teszi, például Ehlers Danlos szindróma vagy Marfan-szindróma .
FISIOPATOLGIA: EGYÉB TOVÁBBI RÉSZLETEK
Az Alport-szindrómához kapcsolódó gének hozzájárulnak egy IV-es típusú kollagén variáns kialakulásához, amely elengedhetetlen a következőkhöz:
- A vese glomerulusok megfelelő szűrése a vérrel szemben.
Magyarázat. A vesékben elhelyezkedő vese glomerulusok speciális erek agglomerátumai, amelyek képesek eltávolítani (mintha szűrőként) a vérből származó hulladékokat és vizeletet képeznek.
A IV. Típusú kollagén megváltoztatásával ezek a veseműködések meghiúsulnak a szűrés és a vizelet létrehozásában.

- A belső fül Corti szerve által készített fordítási munka hanghullámok idegimpulzusokban.
Magyarázat. A Corti orgona a belső fül szerkezete, amely felelős a fül által érzékelt hanghullámok "olvasható" és "értelmezhető" idegjelekké történő átalakításáért az emberi agyból.
A IV. Típusú kollagén módosítása a Corti szervével együtt aláássa a hang transzlációs folyamatát az emberi idegrendszer nyelvébe.
- A lencse megfelelő formájának megtartása, a szaruhártya megfelelő merevsége és a retina pigmentepitéliuma normál színe.
Magyarázat. A kristálylencsék alakja és a szaruhártya merevsége elengedhetetlen a vizuális működéshez és a szem egészségéhez (a retina pigmentepitéliumának színe viszont nem fontos ezeken a területeken).
A megváltozott IV. Típusú kollagén jelenléte a szemében megváltoztatja a kristályos lencse alakját, a szaruhártya merevségét és a retina pigment epithelium színét.
Mindezek az információk magyarázzák, hogy az Alport-szindróma bizonyos tünetekkel kapcsolatos.
Alport szindróma öröksége
Megérteni ...
- Minden emberi gén két példányban van jelen, az allélok, az anyai eredetű és az apai eredetűek.
- Az öröklött betegség autoszomális domináns, ha előfordul, hogy a gén csak egy példányának mutációja elegendő.
- Az öröklött betegség autoszomális recesszív, ha a gén mindkét másolatának mutációját kell előidézni.
Alport-szindróma 3 különböző öröklési modellt mutat be.
A leggyakoribb öröklési minta szerint (az esetek 80% -a) az Alport-szindróma az X-kromoszómához kapcsolódó ún. Örökletes betegség (pl. Hemofília vagy színvakság), mivel ez a kromoszómán található COL4A5-gén mutációjától függ. szexuális X.
A második leggyakoribb öröklési modell szerint (az esetek körülbelül 10% -a) az Alport-szindróma úgy viselkedik, mint az úgynevezett autoszomális recesszív öröklött betegség, mivel jelenléte miatt egy specifikus mutációra van szükség a COL4A3 gének egyikében és COL4A4, amely a 2. kromoszómán található.
Végül, a kevésbé gyakori öröklési modell szerint (az esetek körülbelül 5% -a), az Alport-szindróma az autoszomális domináns öröklött betegség egyik példája, mivel a COL4A3 és COL4A4 gének egyik alléljában egy specifikus mutáció elegendő annak megnyilvánulásához. .
A CHROMOSÓMÁHOZ KAPCSOLATOS ALPORT SYNDROME X
Az X-kapcsolt Alport-szindróma a férfiaknál a legrosszabb hatású.
Ez a jelenség a férfiakra és a nőkre jellemző szexuális kromoszóma-készletekhez kapcsolódik; a nőben valójában két X-kromoszóma van, amelyek a kettő elleni egyik mutáció jelenlétében segítik egymást (az egészséges egyén többé-kevésbé kompenzálja a megváltozott hibákat); emberekben azonban csak egy X-kromoszóma van (a másik egy Y-kromoszóma), amely mutációnak kitéve nem számíthat más, azonos funkciójú kromoszóma támogatására.
AUTOSOMIKUS FELSZERELÉS ALPORT SYNDROME
Az autoszomális recesszív viselkedésű Alport-szindróma tüneteket és hasonló jeleket okoz mind a férfiak, mind a nők körében.
DOMINANT AUTOSOMIC ALPORT SYNDROME
Az Alport szindróma az autoszomális domináns viselkedéssel azonos férfiakat és nőstényeket érinti.
Tünetek és szövődmények
A vese, a belső fül és a szem megfelelő IV. Típusú kollagén hiánya miatt az Alport-szindróma klasszikusan meghatározza: \ t
- A vesefunkció progresszív és meg nem szüntethető elvesztése,
- A hallási képesség csökkenése e
- Szemelváltozások vizuális hiányokkal kombinálva .
Néhány ritka esetben az Alport-szindrómának olyan kiterjedt hatása van, hogy a fentiekben ismertetett következményekhez hasonlóan a nyelőcső és a tracheobronchiális fa széles körben elterjedt leiomyomatózisa, valamint súlyos vaszkuláris problémák ( aorta-disszekció és hasi vagy mellkasi aorta aneurysma). ).
A vesefunkció elvesztése: tünetek
Előfeltétel: a "vesefunkció progresszív elvesztése" kifejezéssel az orvosok azt tervezik, hogy a vesék és belső szerkezeteik (pl. Vese glomerulusok) fokozatosan csökkennek a szűrési kapacitásuk és a vizelettermelésük.
Alport-szindróma jelenlétében a vesefunkció elvesztésével kapcsolatos tünetek főként a hematuria, azaz a vizeletben lévő vér és a proteinuria, azaz a vizeletben lévő fehérje.
A hematuria viszonylag korai tünet, abban az értelemben, hogy Alport-szindrómás betegekben fiatal korban jelentkezik; a legtöbb esetben csak a mikroszkóppal ( mikroszkópos hematuria ) lehet felismerni.
Ezzel szemben a proteinuria egy későbbi tünet, azaz, amikor az Alport-szindróma előrehaladottabb állapotban van.
Csökkent hallási képesség: részletek
A hallás szintjén az Alport-szindróma részleges süketséget okoz; pontosabban, érzékszervi halláscsökkenést okoz, ami megakadályozza, hogy a beteg magas frekvenciákon hallja a hangokat (nagyfrekvenciás halláskárosodás ).
Az Alport-szindrómában szenvedő személyek hallási problémáinak alakulása a mutációtól függő géntől és az öröklődés típusától függően változik; Tény:
- Ha a mutáció a COL4A5-ben található, a férfiak a gyermekkor vége felé alakítják ki a hallásveszteséget, míg a nőstények azokban a ritka körülmények között, amelyekben érdekeltek, öregkorban alakítják ki;
- Ha a mutáció a COL4A4-ben vagy a COL4A3-ban van, és a betegség autoszomális recesszív típusú, mind a beteg, mind a nőbeteg panaszkodik a halláscsökkenés első tüneteiért a késői gyermekkorban vagy korai serdülőkorban;
- Ha a mutáció a COL4A4-ben vagy a COL4A3-ban található, és a betegség autoszomális domináns, mind a férfi, mind a női betegek idős korban érzékszervi halláskárosodást okoznak.

A még ismeretlen okok miatt az Alport-szindróma néhány beteget megment a hallási problémáktól; Más szavakkal, ismeretlen okokból az Alport-szindróma egyes hordozói nem fejtenek ki érzékszervi halláskárosodást.
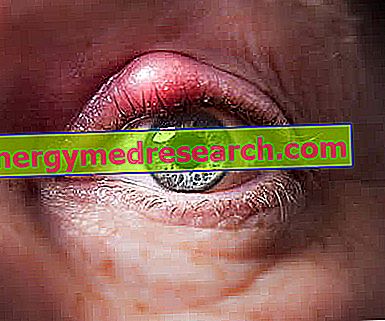
Szemelváltozások és látáshiány: részletek
Az Alport-szindróma által kiváltott szemészeti anomáliák csak a betegek egy bizonyos százalékában megfigyelhetők olyan körülmények között, mint: keratoconus, lenticonous, szürkehályog és foltok jelenléte a retina makulán .
A retina makulán lévő foltok jelenlétének kivételével az összes fent említett állapot többé-kevésbé mélyen érinti a betegek vizuális képességeit.
A nyelőcső és a tracheobronchiális fa diffúz leiomyomatosisa
A nyelőcső és a tracheobronchiális fa diffúz leiomyomatózisa ritka, jóindulatú daganatok, amelyek Alport szindrómában szenvedő betegeknél:
- dysphagia;
- Postprandialis hányás;
- Epigastriás fájdalom és retrosternális fájdalom;
- Ismétlődő hörghurut;
- A nehézlégzés;
- köhögés;
- A lélegzet közben kiabál.
szövődmények
Az Alport-szindróma fő szövődménye a végső stádiumú veseelégtelenség állapota, ami a vesefunkció progresszív és meg nem szüntethető csökkenéséből adódik.
Az orvostudományban a "végstádiumú veseelégtelenség" kifejezés a végső stádiumot, valamint a krónikus veseelégtelenség legsúlyosabb stádiumát jelöli, azaz olyan állapotot, amelyben a vesék teljesen és véglegesen elvesztették minden funkcionális kapacitását.
Nagyon súlyos következményekért felelős (pl. Magas vérnyomás, pulmonalis ödéma, csont törékenység, immunszuppresszió, idegrendszeri károsodás stb.), A végső stádiumú veseelégtelenség krónikus kezelést igényel, mint a dialízis, és a művelet legfontosabb indikációja. sebészeti, mint veseátültetés .
EGYÉB KOMPLIKÁCIÓK
Az Alport-szindróma egyéb lehetséges szövődményei, amelyek szintén magas halálozási arányt mutatnak, a belső vérzés, amely az aortai disszekció vagy esetleges mellkasi vagy hasi aorta aneurysma lebegését követi.
diagnózis
Általában az Alport-szindróma diagnosztizálásához az orvosok a következő adatokat használják:
- A fizikai vizsgálat .
- Az anamnézis ;
- Vesebiopszia ;
- A genetikai vizsgálat .
Fizikai vizsgálat
Ez a beteg által okozott tünetek és jelek orvosi megfigyelése.
Az Alport-szindróma összefüggésében a veseproblémák, halláskárosodások (ha vannak) és a szemészeti anomáliák (ha vannak) egy részét fedi fel.
történelem
Ez a tünetek kritikus vizsgálata specifikus kérdésekkel, a beteg családi történetének vizsgálatával együtt.
Az Alport-szindróma felismerésére szolgáló vizsgálati eljárás részeként az anamnézis lehetővé teszi az orvos számára, hogy megismerje a tünetek megjelenésének pontos pillanatát, és tudja, hogy a beteg családjában van-e örökletes betegség.
Vesebiopszia
A veséksejtek mintájának összegyűjtése és laboratóriumi elemzése.
Az Alport-szindróma összefüggésében hasznos a vesék egészségi állapotának felmérésére és a funkcionális IV-es típusú kollagén hiányának megállapítására.
A vesebiopszia fontos lépés az Alport-szindróma diagnózisában, mert ha időszerű, a lehető leghamarabb megtervezheti a legmegfelelőbb terápiát.
Genetikai teszt
A DNS-elemzés célja a kritikus gének mutációinak detektálása.
Az Alport-szindróma összefüggésében a diagnosztikai megerősítő tesztet és a pontos mutált gén és az öröklődés típusának megállapítását vizsgálja.
terápia
Alport-szindrómából lehetetlen gyógyítani ; az érintettek azonban különböző tüneti kezelésekre támaszkodhatnak, amelyek lehetővé teszik a tünetek szabályozását és a komplikációk kialakulásának elhalasztását.
Tudtad, hogy ...
Annak érdekében, hogy az Alport-szindrómából származó betegségből felépüljön, szükség lenne a genetikai mutáció megszüntetésére vagy a hatások megszüntetésére oly módon, hogy a IV. Típusú kollagén normális és funkcionális legyen.
Tüneti terápia: mit tartalmaz?
Az Alport-szindróma kezelésére szolgáló tüneti kezelések listája a következőket tartalmazza:
- Az ACE-gátlókon alapuló farmakológiai terápia;
- A klasszikus farmakológiai terápiák krónikus veseelégtelenség jelenlétében vannak tervezve;
- dialízis;
- Veseátültetés;
- Hallókészülék használata;
- Keratoconus, lenticonous és / vagy szürkehályog kezelés.
ACE-gátlók
Általában hipertónia és néhány szív- és érrendszeri betegség kezelésére alkalmazzák, az ACE-inhibitorokat Alport-szindróma jelenlétében alkalmazzák, mivel bizonyítottan képesek lelassítani a fent említett örökletes betegségre jellemző vesefunkció progresszív csökkenését.
Ennek köszönhetően az ACE-gátlók lehetővé teszik a veseelégtelenség elhalasztását és a fontos kezelések, például a dialízis vagy a veseátültetés szükségességét.
Ismeretlen okok miatt egyes Alport-szindrómás betegeknél az ACE-inhibitorok hatástalanok; ez magyarázza, hogy az orvosok miért keresnek hasonló erővel rendelkező gyógyszereket.
FARMAKOLÓGIAI TERAPOK A KRÓNIKUS RENÁLIS HIBÁN
Amikor az Alport-szindróma krónikus veseelégtelenséghez vezetett, számos hasznos gyógyszer létezik, köztük:
- A magas vérnyomás elleni gyógyszerek (köztük a már említett ACE-gátlók és ARB-k és a diuretikumok);
- Kalcium- és D-vitamin-kiegészítők (a csontok törése elleni védelem);
- Nátrium-polisztirol-szulfonát és analógok (a kálium felhalmozódásának megakadályozására a vérben).
DIALIZÁLÁS ÉS KIDNEY TRANSPLANTATION
A dialízis olyan kezelés, amely mesterségesen reprodukálja a vese bizonyos funkcióit, megtisztítja a vért a felesleges hulladékból és a vízből.
A veseátültetés viszont egy műtét, amely egy vagy mindkét vesét egy egészséges vesével helyettesíti egy kompatibilis donorból (a donor nem élő vagy élő).
Mind a dialízis, mind a vesetranszplantáció akkor jelezhető, ha az Alport-szindróma a legfejlettebb stádiumban van, azaz a végső stádiumú veseelégtelenségben.
ACOUSTIC APPARATUS
Alport-szindróma jelenlétében a hallókészülék a részleges süketséggel rendelkező betegek számára javasolt gyógyszer.
KERATOCON, LENTICON ÉS KÉSZÍTÉS KEZELÉSE
Amikor az Alport-szindróma a szemeket érinti, keratoconust, lenticonousot és / vagy szürkehályogot okozva, a betegek a szemészeti műtétre támaszkodhatnak, amelyek eredményei több mint kielégítőek.
Milyen orvosi adatokkal jár az Alport-szindróma tüneti kezelése?
Az Alport-szindróma tüneti kezelése több orvosi szakember összehangolt beavatkozását igényli, köztük: gyermekorvosok, nephrológusok, audiológusok és szemészek.
prognózis
Az Alport-szindróma a jelenlétéhez kapcsolódó géntől függően eltérő prognózist mutat; általánosságban azonban soha nem jóindulatú, hiszen a betegség gyógyíthatatlansága egymástól függetlenül, minden betegnek a vesefunkció elvesztését célozza meg.
megelőzés
Az Alport-szindróma lehetetlen a megelőzés feltétele.