Mi a Marfan-szindróma?
A Marfan-szindróma a kötőszövet komplex örökletes rendellenességét írja le, amely elsősorban a szemekre, a szív-érrendszerre és a vázizomrendszerre hat. Tekintettel azonban arra, hogy minden szerv kötőszövetből áll, a Marfan-szindróma ideális esetben elpusztíthatja és befolyásolhatja az egyes anatómiai helyek növekedését és működését.
A szindrómát autoszomális domináns tulajdonságként továbbítják: ezért komoly genetikai betegséggel szembesülünk, amely rendkívül változó fenotípusos expressziót mutat (a hibák nagymértékben különböznek a családtól a betegektől a betegig).
A Marfan-szindróma az FBN1-gén megváltozása (a 15. kromoszómán), amely fibrillin-1-et kódol, egy nagyon fontos kötőglikoproteint, amely a mikroszálak strukturális támogatását képezi.
Mikrofibrilek: fibrillinből állnak, a mikroszálak az extracelluláris mátrixban vannak jelen, amelyben egy rostot képeznek az elasztin lerakódására a rugalmas rostokban. Habár a szervezetben mindenütt jelen van, a mikrofibrilek főként a ciliáris testek aorta, kötőszövetei és övezetei (szemészeti szinten) bővelkednek.
Autoszomális domináns betegségként a Marfan-szindróma csak azokat a gyermekeket érinti, akik örököltek mindkét szülő által megváltoztatott FBN-1 gént. Mindazonáltal a 4-ből egy esetben a betegség spontán mutációk következménye azokban a betegekben, akiknek nincs családi története.
A betegség neve a francia gyermekorvostól származik, aki először 1896-ban írta le (A. Marfan), majd 1991-re kellett várnia, hogy azonosítsa a tüneti megnyilvánulásban szerepet játszó megváltozott gént: a felfedező F. Ramirez volt.
Nézze meg a videót
X Nézze meg a videót a YouTube-onokai
Megemlítettük, hogy a Marfan-szindróma a fibrillin-1-et kódoló gén mutációjának közvetlen expressziója. Próbáljuk meg mélyíteni a témát, hogy megértsük, melyik mechanizmus jön létre a szindróma kiváltására.
A FIBRILLINA 1 az elasztin glikoprotein komponense, amely elengedhetetlen a rugalmasság és a szövet szilárdságának biztosításához és fenntartásához. Fiziológiai körülmények között az 1 fibrillin egy másik fehérjéhez kötődik, amely TGF-béta (vagy transzformáló növekedési béta faktor). Úgy tűnik, hogy a TGF-béta részt vesz a vaszkuláris simaizomot és az extracelluláris mátrixot befolyásoló káros folyamatokban. Ezekből a feltevésekből kiindulva néhány szerző meg van győződve arról, hogy a Marfan-szindróma az FBN-1 gén mutációján túl a TGF-béta feleslegéhez, különösen az aortához, a szívszelepekhez és a tüdőhöz vezet.
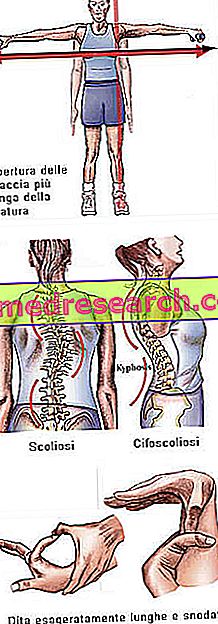
előfordulás
A becslések szerint a Marfan-szindróma 3000-től 5000-ig születik, és a férfiak és a nők között nem egyértelműen jelentkezik, anélkül, hogy a fajta pontosak lennének. A statisztikák azt mutatják, hogy a betegek 75% -ának pozitív családi története van; a fennmaradó 25% -ában az ok a szórványos mutációkban rejlik, amelyek úgy tűnik, hogy valamilyen módon kapcsolódnak az apa előrehaladott korához a fogantatás idején.
A Marfan-szindróma rendkívül súlyos formájú gyermekei várható élettartama kevesebb, mint egy év.
A nyitott szívsebészeti stratégiák kialakulása előtt a legtöbb Marfan-szindrómás beteg átlagos életkora 32 év volt; az orvosi és farmakológiai terápiák folyamatos javulásának köszönhetően jelenleg Marfan-szindrómás betegek átlagosan 60 évig élnek.
Jelek és tünetek
További információ: Tünetek Marfan szindróma
A Marfan-szindróma teljesen tünetmentes lehet. Az érintett betegek túlságosan karcsú szerkezetűek, aránytalanul magasak és vékonyak. Az alsó és felső végtagok sokkal hosszabbak, mint a törzs (dolicostenomegalia). Arachnodactyly- ről is beszélünk, hogy a legjobban kifejezzük az ujjak túlzott hosszúságának fogalmát, ami jellemző a Marfan-szindróma által érintett személyekre: a kezeket tehát összehasonlítjuk a pók lábával.
Ami a magasságot illeti, ezeknek a betegeknek az átlagos magassága a 97. százalékos érték felett van.
A Marfan-szindrómában szenvedő betegeknél gyakran megtalálható egyéb jellemzők:
- A karok nagyobbak, mint a magasság
- Laza ízületek → túlzott ízületi mobilitás
- A mellkasfal deformációja
- Kristályos elmozdulás
- A test felső része kevésbé fejlett, mint az alsó rész
- Spontán pneumothorax (11%)
- scoliosis
- Bőr striae a combon, háton, deltoidon, pectoralis szinten
A Marfan-szindrómához kapcsolódó leginkább problémás jelek közül említjük meg a szívszelep-prolapszist és a mitrális szelep elégtelenségét: hasonló állapotban könnyen előmozdítható az aorta gyűrű és az aorták szétválasztása.
A táblázat a Marfan-szindrómában szenvedő betegeknél előforduló jeleket mutatja. Az ott leírt karakterek nem mindig jelen vannak, de ezek jó része megtalálható.
| Érintett anatómiai hely | Lehetséges tünetek |
csinos | Strawe a mellkasi, lumbális és szakrális területen |
Szem | A látás, az asztigmatizmus, a retina leválás, a keskeny szögű glaukóma, a lencse diszlokáció, a rövidlátás \ t |
Csontszerkezet | Artralgia, kyphoscoliosis, dolicostenomelia (a végtagok túlzott fejlődése a törzshöz képest), hypermobilitás, magas szájpad, deformált mellkas, lapos lábak, keskeny és vékony csuklók, rendellenes visszatérés / szegycsont kiemelkedés, skoliozis, ívelt vállak, spondylolisthesis |
Dita | arachnodactyly |
tüdő | Spontán pneumothorax, dyspnea, idiopátiás tüdő obstruktív betegség |
Arcváltozások | Szájnyálkahártya (szájpadlás), mandibularis retrognathia (az állkapocs kialakulásának hibája), hosszúkás arc |
szív | Angina pectoris, hasi aorta aneurysma, szívritmuszavar, mellkasi aorta dilatáció / szakadás / disszekció, aorta elégtelenség, mitrális szelep prolapsus |
nyelv | Nyelvi nehézség |
diagnózis
Tekintettel a több mint 200 lehetséges mutációra, a genetikai markerek alkalmazása szinte lehetetlen diagnosztikai célokra.
A Marfan-szindróma felismerése nem mindig olyan közvetlen, mert a mutáció fenotípusos expressziója nem mindig nyilvánvaló és könnyen azonosítható. A diagnosztikai késleltetés komolyan veszélyeztetheti a beteg túlélését: csak gondoljunk például arra, hogy nem ismerjük fel a kardiovaszkuláris problémát.
A Marfan-szindróma diagnosztikai kritériumait 1996-ban nemzetközi szinten dolgozták ki: a diagnózis a szindróma fő és kisebb indikátorainak kombinációjával összefüggő családtörténet vizsgálatát tartalmazza.
Néhány használt diagnosztikai teszt a következők:
- echocardiogram
- Mágneses angiorisonancia és CT (az aorta vizsgálatához)
- mágneses rezonancia angiográfia (MRA) kontrasztfolyadékkal (az aorta belső szerkezeteinek nyilvánvalóvá tétele)
- vizsgálata réslámpákkal (a lencse lehetséges eltolódásának elemzése)
- a szemnyomás mérése (a glaukóma lehetséges jelenlétének kiemelése)
- genetikai vizsgálatok (ajánlott a gyermek befogadása előtt, hogy megállapítsa a szindrómát, vagy sem)
terápiák
Genetikai betegségként nincs olyan gyógyszer vagy kezelés, amely megfordíthatja a betegséget.
A gyógyszerek használata azonban elengedhetetlen a tünetek enyhítéséhez és a szövődmények, különösen a szívbetegségek elkerüléséhez. Erre a célra különösen alkalmasak a vérnyomáscsökkentő gyógyszerek, például a szartánok (különösen), az ACE-gátlók és a béta-blokkolók.
A Marfan-szindróma összefüggésében a scoliosisban szenvedő betegek speciális ellátást követhetnek, valamint a glaukóma által érintettek számára.
A műtét elképzelhető, hogy korrigálja az anomális aorta dilatációt, ami gyakran összekapcsolja a Marfan-szindrómás betegek többségét.
Folytatás: Marfan szindróma - Kábítószerek és gondozás »


